







Syndromes in ENT

Alpert Syndrome
AKA: hereditary nephritis
-
kidney disease (hematuria, edema, and hypertension)
-
loss of hearing (bilateral sensorineural hearing loss) Hearing loss becomes apparent in late childhood or early adolescence, usually before the onset of kidney failure, and starts with high-frequency loss.
-
eye abnormalities (cone-shaped lens (anterior lenticonus)
-
Diffuse leiomyomatosis (smooth muscle overgrowth in the respiratory and gastrointestinal tract) dysphagia, postprandial vomiting, recurrent bronchitis, substernal or epigastric pain, dyspnea, stridor, and cough.
Pathophysiology:
impaired production and deposition of collagen
4α345 network in the basement membranes of the glomerulus, cochlea (inner ear), and eye.

Ramsay Hunt Syndrome
AKA: Herpes Zoster Oticus / geniculate ganglion herpes zoster
-
ipsilateral facial paralysis
-
otalgia
-
vesicular rash
Pathophysiology:
varicella-zoster virus, a member of the human herpesvirus family. Once the clinical VZV infection, chickenpox, has cleared, the virus remains latent in cranial nerves or dorsal root ganglia and may subsequently reactivate in times of physiological stress or immunocompromise, leading to herpes zoster, known as "shingles" anywhere on the body or "Ramsay Hunt syndrome" when facial paralysis is involved

Sjögren's syndrome
-
Xerostomia (Dry mouth)
-
Keratoconjunctivitis sicca (bilateral desiccation of the conjunctiva and cornea due to an inadequate tear film)
-
Swelling of exocrine glands (sweat glands, lacrimal glands, salivary glands, mammary glands, and digestive glands in the stomach, pancreas, and intestines)

Lip Biopsy for Sjogren's Syndrome (Minor Salivary Gland Biopsy)

Grisel Syndrome
Torticollis : Tilted head & limited range of motion
Nontraumatic atlantoaxial subluxation (AAS) is a rare complication of upper respiratory tract infections. Grisel’s syndrome refers only to non-traumatic AAS, and most commonly occurs after an upper respiratory infection, though some unusual causes include mumps, tuberculosis, and Kawasaki. It may also be seen after certain otolaryngologic operations such as adenoidectomies, tonsillectomies, and ear surgeries.
TYPES OF MENIERE'S DISEASE
1. Classical Meniere's disease
2. Vestibular Meniere's disease - vestibular
symptoms and aural pressure
3. Cochlear Meniere's disease - cochlear symptoms
and aural pressure
4. Lermoyez syndrome Reverse Meniere's
5. Tumarkin's crisis - Utricular Meniere's
Lermoyez Syndrome
Tinnitus and loss of hearing prior to an attack of vertigo, after which hearing improves. It is considered a variant of labyrinth idropsy, similar to Ménière’s disease. While Ménière's disease is chronic and progressive, Lermoyez's syndrome is an acute phenomenon which results in no damage for the ciliated cells of cochlea. This is possible because the excess of pressure inside the inner ear is reduced by a break of the sacculum, in the vestibular labyrinth, with no long term consequence for the cochlea.


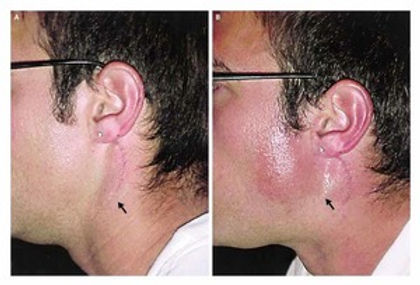
Frey syndrome
AKA: Baillarger’s syndrome / auriculotemporal syndrome / Dupuy syndrome/ gustatory hyperhidrosis
Present with facial warmth, flushing, and sweating in the territory of the auriculotemporal nerve overlying the parotid gland, which may include the preauricular skin, the temporal skin, the scalp, and the temporomandibular joint region
Pathophysiology
When an insult to the parasympathetic and sympathetic nerve fibers of the auriculotemporal nerve in the parotid region occurs, the resulting aberrant regeneration of post-ganglionic parasympathetic nerve fibers (responsible for salivary secretion) along the pre-existing sympathetic pathways to the vessels and sweat glands of the skin leads to the development of Frey syndrome.

Pierre Robin Sequence
Pierre Robin Sequence is a congenital group of defects resulting in upper airway obstruction and failure to thrive. It can be syndromic or occur in isolation.
(
It is called a "sequence" because a single developmental defect results in a chain of secondary defects. More specifically:
-
Defect 1: Micrognathia due to mandibular hypoplasia
-
Defect 2: Glossoptosis due to posterior displacement of the tongue
-
Defect 3: U-shaped cleft palate due to blocked closure of the palatal shelves
A small jaw results in the tongue occupying more significanter proportion of the oropharynx and falling back to cause airway obstruction.

Down Syndrome
1. Small ears.
2. Narrow external ear canals: can make it diffcult to view the tympanic membrane or insert grommets.
3. Increased incidence of glue ear: age related.
4. Otoacoustic emissions often non-reproducible, even in patients with normal hearing ability.
5. Abnormal ossicles.
6. Hearing loss: Mixed (7%) Sensorineural deafness (8%).
7. Airway obstruction from small posterior nasal spine with large tonsils and adenoids.
8. Macroglossia.
9. Subglottis: usually require one anesthetic tube size smaller than expected.
10. Unstable neck, with a risk of atlanto-occipital joint subluxation in 15%, especially during general anesthetic

CHARGE Syndrome
•Coloboma
• Heart defect
• Atresia choanae
• Retardation of growth and development
• Genital defect and ear anomalies and/or deafness
Mutations in the gene CHD7 can be identifed in approximately 65% of cases.
ENT features
1. Microtia: Usually unilateral or bilateral.
2. Pre-auricular skin tags.
3. Deafness: sensorineural and/or conductive.
4. Middle ear: ossicular malformations.
5. Temporal bone abnormalities: absent or hypoplastic semicircular canals.
6. Macrostomia: occasionally with lateral facial clefting. 7. Choanal atresia/stenosis (unilateral or bilateral)

Usher Syndrome
• Most common cause of deafness and blindness among pediatrics representing 10% of hereditary deafness
Hearing loss
vestibular deficits
ataxia
retinitis pigmentosa (RP)
• Type I: most common (90%), profound deafness, RP by age 10, absent vestibular response
• Type II: moderate/severe deafness, RP by teens/twenties, normal or slightly decreased vestibular response
• Type III: progressive HL, RP begins with puberty
• Type IV: X-linked; clinically similar to type II


Branchio-Oto-Renal Syndrome
AKA: Melnick–Fraser Syndrome
An association of:
1) branchial fistulae or cysts
2) ear malformations, which can include the inner, middle and outer ear
3) renal malformations, which can range in severity from renal hypoplasia to agenesis.
ENT features:
1. Ear anomalies: pits in the pre-helical region and dysplastic pinnae.
2. Conductive (due to ossicular malformations), sensorineural,or mixed hearing impairment. Inner ear malformations may include Mondini dysplasia of the cochlea, and occasionally, dilated vestibular aqueducts could be found.
3. Branchial fstulae and/or cysts
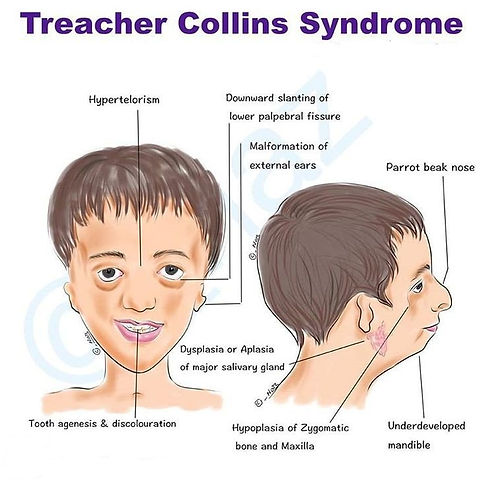
Treacher Collins Syndrome
A genetic syndrome with bilateral abnormalities of the first and second branchial arches combining Tessier cleft numbers 6-9.
Sporadic or autosomal dominant inheritance of variable penetrance. TCOF1 is the commonest genetic mutation.
1. Microtia, dysplastic ears (bilateral ear anomalies).
2. Auricular pits/fstula/tags.
3. Deafness: usually conductive due to external auditory canal atresia and malformations of the ossicles.
4. Cleft palate.
5. Micrognathia: cleft palate and micrognathia together may result in signifcant airway problems in neonates.
6. Mandibular hypoplasia.
7. Flat malar region.
8. Prominent nose.
9. Broad mouth.
10. Narrow nasopharynx.
11. Choanal atresia (rare).

22q11.2 Deletion Syndrome
AKA: velocardiofacial syndrome/ DiGeorge syndrome/ Shprintzen syndrome.
ENT Features
Palate
1. Overt or submucous cleft palate.
2. Velopharyngeal incompetence.
3. Hypernasal speech.
Ears
1. Small, low-set ears. \
2. Prominent/over folded helices.
3. Absent or hypoplastic ear lobules.
4. Pre-auricular tags.
5. Otitis media.
6. Sensorineural hearing loss.
7. Narrow external auditory canals


Pendred Syndrome
combination of sensorineural hearing loss and thyroid goiter with or without hypothyroidism.
• SNHL associated with iodine metabolism defect leading to euthyroid goiter
• Associated with Mondini’s dysplasia (cochlea with incomplete partitioning and a reduced number of turns, an enlarged vestibular aqueduct and a dilated vestibule) and enlarged vestibular aqueduct
• Historically diagnosed with perchlorate discharge test
Palato-Pharyngo-Laryngeal Rhythmic Myoclonus in Neuro-Bechet Syndrome
Behcet syndrome

Behcet disease is an auto-inflammatory systemic vasculitis of unknown etiology. It is characterized by mucocutaneous manifestations, including recurrent oral and genital ulcerations, ocular manifestations, especially chronic relapsing uveitis, and systemic vasculitis involving arteries and veins of all sizes. It is also known as Behcet syndrome and malignant aphthosis.